No ano de 1866, Laurence e Moon descreveram uma condição genética na qual os indivíduos apresentam atraso mental, baixa estatura, hipogenitalismo, ataxia, paraplegia espástica e nistagmo. Outros também exibiam retinose pigmentar ou atrofia coroidiana. Anos depois, Bardet e Biedl descreveram uma síndrome composta por obesidade, polidactilia, retinose pigmenar, atraso mental e atresia anal. Tempos depois, Solis-Cohen e Weiss uniram as duas desordens, nomeando-a de Síndrome de Laurence-Moon-Biedl. Contudo, mais recentemente, foi estabelecido que as síndromes de Laurence-Moon e Bardet-Biedl tratam-se, na verdade, de patologias distintas.
A síndrome de Bardet-Biedl é definida como uma alteração genética, de caráter autossômico recessivo, caracterizada por distrofia retiniana, polidactilia, obesidade, atraso mental e hipogenitalismo. Estima-se que esta síndrome tem uma prevalência que varia entre uma pessoa de 17.500 até uma entre 160 mil.
Mutação em no mínimo 14 genes distintos pode levar à síndrome de Bardet-Biedl. Acredita-se que estes genes estão desempenham papéis importantes nas estruturas celulares denominadas cílios, que são estruturas presente na superfície de diferentes tipos de células, e responsáveis pelo movimento celular. Além disso, os cílios também são essenciais na percepção de estímulos sensoriais, como a visão, audição e olfato. Desta forma, defeitos nos cílios, resultam em alterações de todas essas funções.
Mutações nos genes BBS levam à síndrome em questão. Aproximadamente 25% dos casos da síndrome são decorrentes de uma mutação no gene BBS1, enquanto que 20% dos casos são ocasionados por mutações no gene BBS10. Mutação em cada um dos outros genes BBS representa uma pequena porcentagem de todos os casos da doença.
Uma das principais manifestações clínicas é a perda de visão, por causa de alterações na retina. Esta perda começa ainda na infância, primeiramente com a perda da visão noturna, que evolui para pontos cegos da visão periférica. Com o tempo, vai acontecendo a ampliação desses pontos cegos, que leva a uma considerável perda da visão, ou até mesmo a perda completa da visão.
Obesidade é outra característica desta síndrome. O excessivo ganho de peso inicia-se, geralmente, na infância, passando a ser um problema para o resto da vida do indivíduo. Em associação com a obesidade, pode haver diabetes do tipo II, hipertensão, elevados níveis de colesterol e hipercolesterolemia.
Outros sintomas e manifestações clínicas desta síndrome incluem:
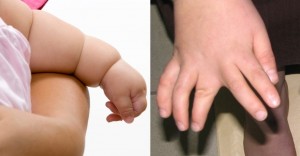
– Polidactilia – dedos extras nas mãos ou nos pés;
– Atraso mental – metade dos portadores da síndrome sofrem com atrasos mentais;
– Anormalidades da genitália – os homens têm os órgãos sexuais menores e as mulheres tem ciclo mestrual irregular;
– Anormalidades renais – podem levar a insuficiência renal;
– Atraso no desenvolvimento das habilidades motoras – muitas vezes é consequência da obesidade;
– Dedos anormalmente curtos e/ou fundidos nos pés – peles que ligam um dedo ao outro;
– Perda parcial ou completa do olfato;
– Alterações hepáticas, cardíacas e gastrointestinais.
O diagnóstico é clínico, havendo também a opção de teste molecular para 4 dos 14 genes envolvidos na síndrome identificados até o momento.
O tratamento é somente em relação aos sintomas, porque não existe um tratamento para todos os sintomas presentes nesta síndrome e também não há uma cura definitiva para a doença.
Fonte: InfoEscola e Retina Brasil